




生命體中的蛋白質結構在不斷變化。預測蛋白質結構的動態變化,對理解生命過程,研發新型藥物都有著重要的意義。
西湖大學李子青團隊與廈門大學、德睿智藥合作,首創研發了能夠刻畫蛋白質構象變化與親和力預測的AI模型ProtMD。這是第一個嘗試解析蛋白質動態構象的AI方法,可輔助藥物化學專家更加精準的篩選出高活性小分子,從而加速臨床前藥物研發。相關研究成果發表在Advanced Science。

19世紀Fischer提出的鎖鑰學說認為,蛋白和小分子在結合過程中是剛體,并不會發生任何形變;而當今的生物學家早已放棄了這一論斷,他們發現蛋白不僅會有形變,而且有隨機的抖動和跳躍。
1965年諾貝爾物理學獎的獲得者,理查德·費恩曼(Richard Feynman),曾做過一個很有名的論述:”如果一定要給一個最重要的假設,來幫助科學家們理解生命,那就是萬物皆由原子構成,一切生命體可以理解成原子的跳躍和抖動。”在他眼里,根據第一性原理,原子的運動是生命的起點。過去五十年的生物物理學都在致力于更好地理解原子運動的機理。
過去,科學家通過計算機模擬藥物分子和靶點蛋白的相互作用,篩選出高靶點親和力的藥物分子,然而由于靶點蛋白的三維構象在不同生理環境下具有一定的隨機性,因此直接使用靶點蛋白質的靜態結構與分子對接,可能導致預測結果的嚴重偏差。
現在,DeepMind研發的AI方法AlphaFold2,能夠準確預測蛋白質的三維結構,對結構生物學、藥物設計、乃至整個科學界都產生了巨大影響。但AlphaFold2只能預測蛋白質在一個瞬間的靜態結構,尚未能解決蛋白質結構動態變化的預測。
然而,預測蛋白質結構的動態變化,對理解生命過程、研發新型藥物都有著重要的意義。尤其在AI藥物設計中,通過對藥物分子與靶點蛋白結合后的動態結構變化的預測,評估藥物-靶點結合親和力和藥物效果,是提高AI藥物篩選準確性和效能的重要思路。
李子青團隊首創開發了預測蛋白質結構動態變化的AI模型ProtMD。給定藥物分子和靶點蛋白,ProtMD預測藥物分子與生物體內靶點蛋白質結合(柔性對接)后蛋白質結構的變化過程,推斷藥物與靶標蛋白結合的穩定性,預測藥物功能,從而提升AI藥物設計的精度和效率。
讓我們來復盤一下李子青團隊的動態構象研究方法,包括數據生成與AI建模兩個環節。
(1)用分子動力學生成蛋白質“跳動”的軌跡數據
過去,基于牛頓力學確定論的熱力學計算方法,通過模擬分子體系的運動可以計算出蛋白質的動態序列,生成蛋白質“動”的數據。雖然該方法運算量巨大,耗時較長,但該方法所生成的數據,正好可以用于訓練AI模型。
研究團隊從蛋白質數據庫PDB中共計57651個人類蛋白結構中,選取了具有代表性的數十個蛋白質結構,使用Molecule Dance(分子跳動)平臺對這數十個蛋白質進行分子動力學模擬——團隊采用對蛋白質動態結構“抓拍”的序列,建立蛋白質動態構象的模型,就像人們用靜態照片序列來形成動態視頻一樣,獲得了數TB大小的蛋白質的空間運動軌跡,作為AI建模的依據。
(2)用AI方法對蛋白質構象變化進行建模
解決思路是這樣的:第一,算法需要能夠基于上一時刻的蛋白的“樣子”,預測下一時刻的蛋白變成什么樣;第二,即使把時間順序打亂,算法能夠將根據蛋白質長的“樣子”按照正確的時間順序重新排序。
為實現這樣的目標,研究團隊在傳統NLP和CV預訓練方法的基礎上,為ProtMD建模創新設計了兩個對應的自監督學習任務。第一,要求ProtMD模型能夠基于上一時刻的蛋白構象預測下一時刻的蛋白構象。第二,訓練ProtMD模型對不同時刻蛋白質順序的排序能力,使其能對時序被隨機打亂的蛋白質構象進行排序。完成訓練后,ProtMD即可預測藥物分子與靶點蛋白結合后的構象變化,以評估藥物效果(如下圖)。
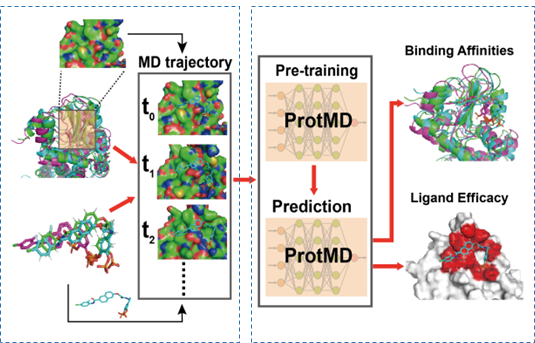
圖.構象變化軌跡建模(左框)和模型用于藥物分子親和力預測和配體功效預測(右框)
實驗表明,ProtMD在藥物-蛋白親和力預測任務上,輕量級版本表現已超過現有的最優(SOTA)模型。在配體功效預測任務上,ProtMD重量級版本AUPRC較SOTA模型提升14%。ProtMD的表現不僅說明該模型的能力,而且證明引入蛋白質時空動態信息,可顯著提升藥物親和力預測準確性,輔助藥物化學專家更加精準的篩選出高活性小分子。
這項研究是采用AI方法解析蛋白質動態構象邁出的第一步。李子青認為,傳統的藥物蛋白結合理論基于靜態蛋白構象,而實際上蛋白構象在藥物結合之前和之后是會發生變化的。ProtMD是預測蛋白-藥物結合過程中動態構象的一個嘗試,使得AI藥物設計能夠更準確地完成藥物-蛋白親和力預測這一核心任務,從而提升AI藥物設計的有效性。德睿制藥CEO牛張明博士認為,ProtMD的研發為基于蛋白的機器學習預訓練模型研發提供了新方向。該方法在底層原理上實現了突破,在實驗預測精度上超過“同類最優”,證明了蛋白質動態時空信息在蛋白-小分子親和力預測上的重要性。ProtMD的工業級版本可大幅提高藥物親和力預測與虛擬篩選效率。
西湖大學李子青實驗室科研助理吳方、廈門大學博士生金淑婷、德睿制藥AIDD總監江熒輝為本文共同第一作者,西湖大學AI講席教授李子青(Stan Z.Li)為本文通訊作者。本項目得到了國家科技部“新一代人工智能”重大項目和國家自然科學基金重點項目的支持。
西湖大學聚焦基礎前沿科學研究,致力尖端科技突破,注重學科交叉融合,努力實現原始創新和科技成果轉化的重大突破。深耕AI領域的李子青,2019年加入西湖大學,取得了多項AI+交叉學科研究成果:與郭天南實驗室合作,創新基于AI的蛋白質生物標志物發現及其臨床診斷應用[1][2],成果已產業化;與李凌實驗合作,創新了AI天氣預測新方法[3];在AI蛋白質序列設計的精度和速度上也取得了目前最好的性能[4]。
References
[1]Sun,Yaoting,et al."Artificial intelligence defines protein-based classification of thyroid nodules."Cell discovery 8.1(2022):1-17.
[2]Zhang,Fangfei,et al."Phenotype classification using proteome data in a data-independent acquisition tensor format."Journal of the American Society for Mass Spectrometry 31.11(2020):2296-2304.
[3]Lin,Haitao,et al."Conditional local convolution for spatio-temporal meteorological forecasting."Proceedings of the AAAI Conference on Artificial Intelligence.Vol.36.No.7.2022.
[4]Gao,Zhangyang,Cheng Tan,and Stan Z.Li."PiFold:Toward effective and efficient protein inverse folding."arXiv e-prints 2022:https://arxiv.org/abs/2209.12643.